Ending Your Patients’ Diagnostic Odyssey via Maximized Coverage With Enhanced Whole Genome Sequencing
Have You Ever Heard of Gaucher Disease?
Gaucher Disease (GD) is a rare genetic disorder that results in the body’s inability to produce an essential recycling enzyme. This deficit can cause a range of symptoms varying in severity – making it difficult to diagnose. Without a clear diagnosis, patients may spend years on their diagnostic odyssey, which ultimately delays access to potential treatments. However, thanks to the advancements of sequencing technology, it is now possible to quickly and comprehensively diagnose GD and many other genetic disorders. Keep reading to learn more about GD and how Whole Genome Sequencing (WGS) can help end your patients’ diagnostic odyssey.
GD is a Lysosomal Storage Disease (LSD) caused by pathologic variants in the GBA1 gene. This gene sequence encodes an enzyme called β-glucocerebrosidase, which recycles large molecules present in cell membranes.1
The onset of GD signs and symptoms can happen anytime, and some people are not diagnosed until they are adults.2 Variants in the GBA1 gene have also been found to increase the risk of developing PD. 5–20% of patients with PD can be estimated to carry variants in the GBA1 gene. Thus, associating the GBA1 gene variants with common complex disorders, like PD, increases interest in analyzing patients using WGS.3
Signs & Symptoms
First described in 1882 by Dr. Philippe Gaucher, this autosomal recessive disease is divided into three types:
Gaucher Disease Type 1
Type 1 accounts for almost 90% of all cases and is common among Ashkenazi Jews. With Type 1, there is no primary central nervous system involvement, but bone disease and other symptoms, such as the below, may be experienced:
Splenomegaly
Hepatomegaly
Cytopenia 2
Pulmonary disease
Gaucher Disease Type 2
Type 2 is characterized by an early onset and is usually fatal by the age of 2. If a patient has type 2 GD, no bone disease will be present; however, they will likely experience primary central nervous system involvement, often in the form of the below symptoms:
Type 3 affects the nervous system more severely than types 1 and 2. The primary central nervous system is often affected and presents with the below symptoms:
Oculomotor apraxia
Seizures
Progressive myoclonic epilepsy
Patients will also often have none disease and other symptoms, such as:
Some Patients May Develop Physical and Mental Conditions:
Physical
Delayed growth and puberty
Jaundice (yellow coloration of the skin and whites of the eyes)
Hepatosplenomegaly (enlarged liver and spleen)
Anemia (low levels of red blood cells)
Asymptomatic osteopenia (decrease in bone mineral density)
Parkinsonism
Behavioral, Social, and Emotional
Depression and anxiety
Difficulty coping with the disease and its effects on daily life (adjustment disorder)
Social isolation and withdrawal
Anhedonia (decreased interest in hobbies and activities)
Reduced quality of life
Speech and Language
Dysarthria (difficulty with articulation and pronunciation)
Delayed speech and language development
Stuttering (speech fluency disorder)
Aphasia (difficulty understanding and using language)
Intelligence and Learning
Learning difficulties
Cognitive impairment
Decreased attention and concentration
Dementia
Did You Know?
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer's Disease (AD) – affecting over 10 million people worldwide from all walks of life. Susceptibility to the development of the more common late-onset forms of PD disease has been associated with variants in several genes, including GBA1.
How Can Gaucher Disease Be Diagnosed and What Are the Challenges?
Measuring the activity of the glucocerebrosidase in macrophages and identifying pathogenic variants in the GBA1 gene are commonly used methods to diagnose GD.4
As is often the case, the method of identifying these variants depends on the patient’s medical history. While patients with clear clinical profiles can be diagnosed relatively easily using single gene testing (e.g., the Sanger Method), diagnostic challenges may arise for patients with heterogenic phenotypes and unclear or atypical symptoms. These patients with prior unsuccessful testing require a rapid diagnosis in order to initiate the required treatment.
Identifying variants in gene coding regions has been the focus of most studies on genetic diseases, but this only accounts for approx. 1–2% of a patient’s entire genome.
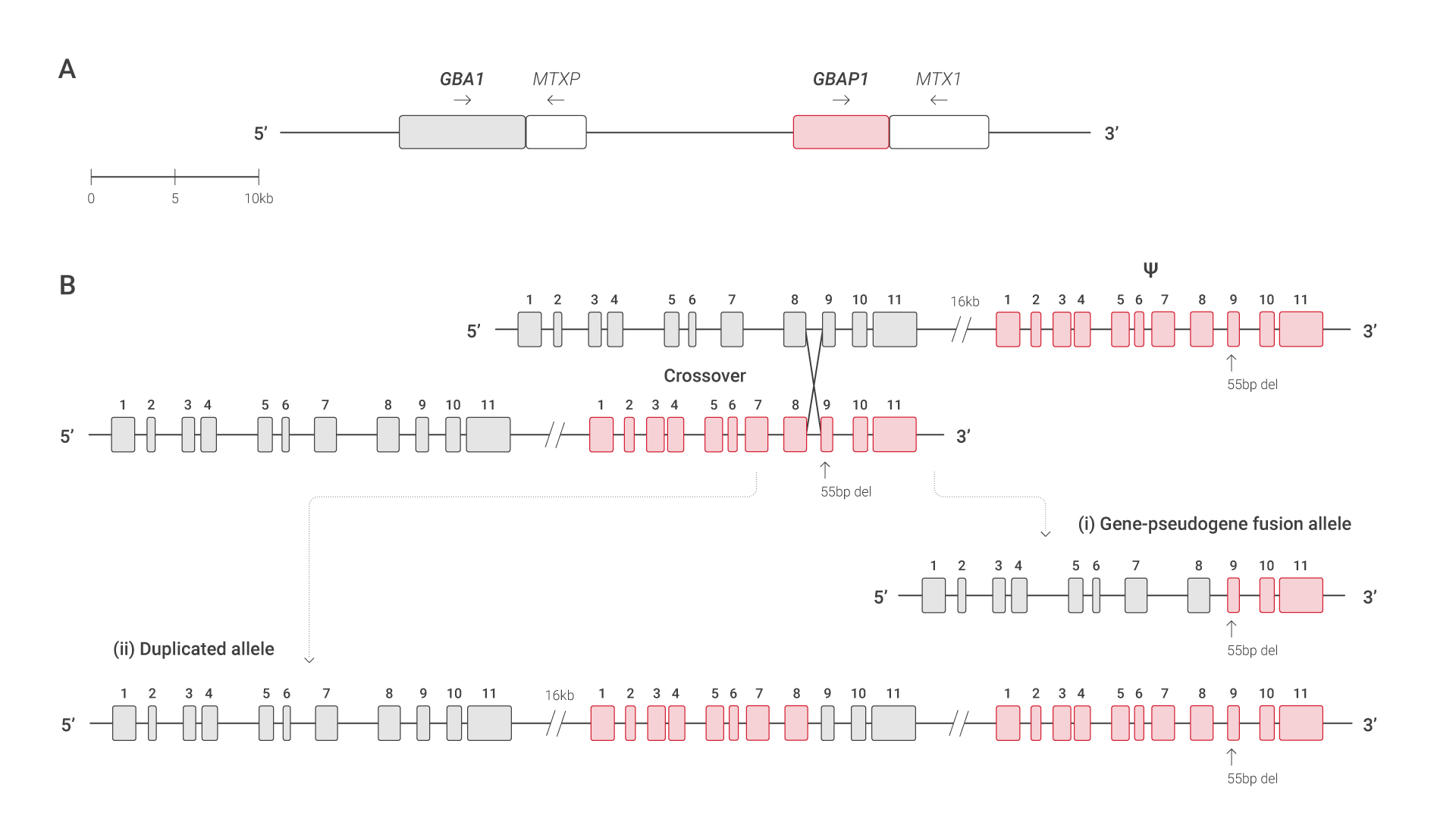
In the case of GD, matters are even more complicated, with the presence of non-functional GBAP1 pseudogene sharing high sequence similarity with the functional GBA1 gene – making it difficult to carry out molecular analyses.
Recombinant alleles are especially difficult to detect using NGS, as a reciprocal crossover event may result in a gene-pseudogene fusion allele, which can lead to false negative results due to the presence of the GBAP1 pseudogene (see figure).5
Consequently, there is a need for more comprehensive analyses that can detect key pathogenic variants located outside gene coding regions, including the non-functional GBAP1 pseudogene.
The implementation of genomics in clinical praxis is thus increasing, and the number of supporting studies is growing. WGS has become the preferred method, as it provides a more comprehensive analysis and increases the chance of molecular diagnosis.
Professional society guidelines recommend WGS as an option for a first-line test (e.g., the American College of Medical Genetics (ACMG) and the European Society of Human Genetics (ESHG)), and adoption of WGS in national health systems (e.g., in the UK and France) has begun. Better affordability and increasing demand for early and rapid diagnosis are driving the growth of this next generation of sequencing.
A Scaled representation of the GBA1 gene (gray) with and their highly homologous GBAP1 pseudogene (red) B Example of a reciprocal crossover event, resulting in a
(i) gene-pseudogene fusion allele and a (ii) duplicated allele.
If the sequence containing the pseudogene-derived segment (red) of the recombinant alleles is read, the pseudogene could imply a false-negative result.
With NEW CentoGenome, we introduced a tool enabling the accurate detection of these recombinants and improving the diagnostic approach, to provide Gaucher patients with the life-changing answers they need.
Maximized Coverage With Whole Genome Sequencing
As you can see, diagnosing GD can be challenging, especially in cases with atypical symptoms or heterogenic phenotypes. However, with an enhanced sequencing approach that captures the complete clinical picture, the chances of identifying pathogenic variants located outside gene coding regions, including the critical GBAP1 pseudogene, are significantly improved.
CENTOGENE’s newly enhanced WGS service, NEW CentoGenome, integrates the latest methods to enable maximized coverage to diagnose genetically complex and undiagnosed cases with the highest level of certainty. NEW CentoGenome can detect almost all changes in a patient’s DNA by sequencing both the entire protein-coding and the non-coding regions of the genome. This also allows for the identification of the critical GBAP1 pseudogene, whose recombination with GBA1 genes causes 12% of the disease-causing alleles – significantly supporting the diagnosis of patients with heterogeneous phenotypes and unclear or atypical symptoms.
With proper diagnosis, patients can receive the appropriate therapeutic treatment, such as Enzyme Replacement Therapy (ERT), which can significantly reduce GD-related symptoms and disease complications.6
Gaucher Disease – It’s covered with NEW CentoGenome!
Do You Want To Learn More About Gaucher Disease and Whole Genome Sequencing?
Don’t miss out on the opportunity to learn from our experts! Register now for our upcoming webinar with Dr. Aida M. Bertoli-Avella, CENTOGENE’s Head of Research Data Analysis and Dr. Tobias Böttcher CENTOGENE’s Director Clinical Neurogenetics, and discover how you can successfully diagnose and treat complex cases. During the webinar, you’ll hear about our experience in ending the diagnostic odyssey of one of our patients and learn about the latest advances in genomics and diagnostic techniques. Register today to secure your spot and take the first step in improving patient outcomes with NEW CentoGenome.
Webinar on demand
The Power of Whole Genome Sequencing: Uncovering New Variants for Precision Medicine
3. Woo, E. S., Tayebi, N., & Sidransky, E. (2021). Next-Generation Sequencing Analysis of GBA1: The Challenge of Detecting Complex Recombinant Alleles. Frontiers in Genetics, 12. https://doi.org/10.3389/fgene.2021.684067
5. Woo, E. S., Tayebi, N., & Sidransky, E. (2021). Next-Generation Sequencing Analysis of GBA1: The Challenge of Detecting Complex Recombinant Alleles. Frontiers in Genetics, 12. https://doi.org/10.3389/fgene.2021.684067